
FDA 510(k) Approval: What is it, and why is it important?

Apr 9, 2025
Introduction
In a market flooded with air purifier options, obtaining 510(k) approval from the Food and Drug Administration (FDA) can offer a significant competitive advantage. This approval enhances the air purifier's credibility and trustworthiness among consumers, healthcare providers, and company stakeholders by indicating that the product has been rigorously tested and meets high standards. Official recognition and approval by a regulatory authority can be a decisive factor for buyers. It simplifies entry into international markets, as many countries recognize or require FDA approval as part of their regulatory processes, facilitating global distribution.
Overview of 510(k) testing
The 510(k) premarket notification is a regulatory process required by the FDA for specific medical devices, including stand-alone and HVAC system air purifiers, that help reduce pathogens in healthcare settings. This process ensures that new devices are at least as safe and effective as legally marketed devices (predicates) that are not subject to premarket approval.

Testing Species: Standard testing for a 510(k) involves challenging an air purifier with every species type the company wants to claim their device is effective against. A typical 510(k) test matrix covers testing with unenveloped RNA viruses, unenveloped DNA viruses, gram-positive bacteria, gram-negative bacteria, mold spores, and bacterial endospores. More options for types of species that can be tested include enveloped RNA viruses, fungal bacteria, and mycobacteria. It is important to note that the FDA only allows claims against the species the device tested with. However, broad claims against species types are acceptable.
Testing Matrix: Testing is typically done in triplicate device challenge trials with at least one control trial per test species. For the control trial, the testing parameters remain the same as those used for the test trials, but no device is in operation. Data from this testing is used to calculate the natural decay rate of each species over time in the testing system. Depending on an air purifier's settings and components, the FDA may require a more encompassing test matrix to include multiple speed settings and filterless testing.

Graphical representation of the various filtration types available in air purifiers. Components may need to be tested separately in a 510(k) test matrix.
Testing Results
To calculate the reduction yielded by a test device during 510(k) testing, results from the control trials are plotted on a logarithmic (log) graph to show the natural viability loss of each species over time in the chamber. All data is normalized to time zero concentrations and plotted to show the loss of viability over time. The device is attributed to the reduction after factoring in the control losses (Figure 3). The FDA requires the test device to achieve a 4.0 net log reduction (>99.99%) with any species the company plans to claim against. Note that a 1 log reduction is equivalent to 90%, a 2 log reduction is equivalent to 99%, and so on.
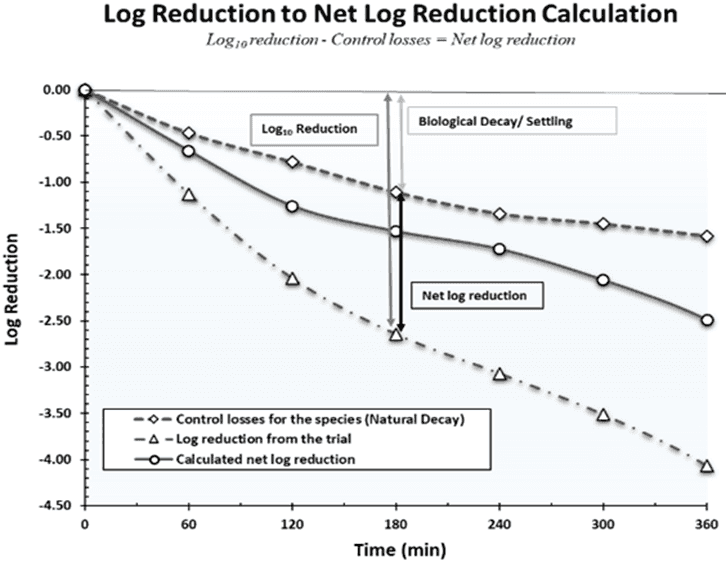
Graph showing log reduction to net log reduction using the controls run during testing.
1.Why is 510(k) Testing Important? Ensures Safety and Efficacy: 510(k) approval requires manufacturers to provide substantial evidence that their air purifier is safe and effective. This rigorous process ensures that the device will perform as intended without harming users.
2. Enhances Credibility and Trust: An air purifier with 510(k) approval has undergone thorough testing and review, enhancing its credibility among consumers, healthcare providers, and other stakeholders. It signifies that the product meets high standards of quality and performance.
3. Facilitates Market Access: Many international markets recognize FDA approval as a benchmark for safety and efficacy. Obtaining 510(k) approval can simplify entering global markets, as many countries have regulatory processes that align with or recognize FDA standards.
4. Creates a Competitive Advantage: Having 510(k) approval in a crowded market can provide a significant competitive edge. It differentiates the product as officially recognized and validated by a reputable regulatory authority, which can be a decisive factor for buyers when choosing various options.
Implications for Public Health and Building Design
The 510(k) approval process is critical in enhancing public health by ensuring the safety and efficacy of air purifiers. Its implications extend to building design, influencing how spaces are constructed and maintained to promote healthier environments. By adhering to these standards, manufacturers, architects, and building managers can create safer, more resilient spaces that protect and enhance the well-being of their occupants.
Additional Air Purifier Testing
Along with FDA 510(k) registration, there are many other tests that air purifier manufacturers can do to gain even more of a competitive advantage in the ever-growing air purifier market. Some of the other testing options to consider for setting an air purifier apart from the rest include:
EPA Registration Testing: Ensuring compliance with environmental safety and performance standards.
ASHRAE 241: Control of Infectious Aerosols
AHAM AC-5: Method for Assessing the Reduction Rate of Key Bioaerosols by Portable Air Cleaners Using an Aerobiology Test Chamber
ASTM E3273-21: Standard Practice to Assess Microbial Decontamination of Indoor Air using an Aerobiology Chamber
References and Further Reading
If you are interested in FDA 510(k) testing or any of the other testing listed above or need more detailed information regarding the testing standards, contact ARE Labs.